
Antibiotic-resistance infections represent a real threat to the quality of healthcare and life expectancy for every country. Recently in 2019, the CDC declared in its report that more than 35,000 people die due to antibiotic-resistant infections. This figure was just a subset of a group of 2.8 Million individuals who were affected by the so-called antibiotic-resistant (AR) infections.
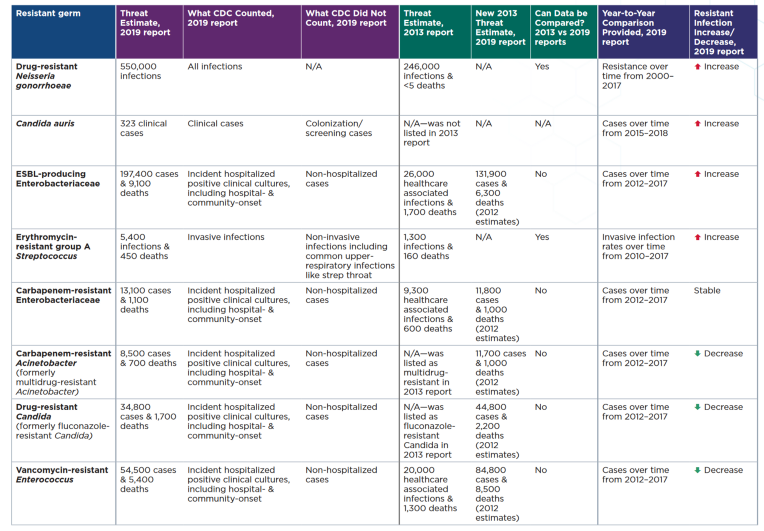 |
|---|
| This table summarizes the 2019 AR Threats Report estimates, and compares these estimates to the 2013 report when applicable. U.S. Centers for Disease Control and Prevention. Return to this paper to learn more |
The report lists 18 antibiotic-resistant germs (bacteria, fungi, and germs). Ten of the 18 antibiotic-resistant threats are Gram-negative, but few antibiotics are available or in development to treat the infections they cause. DRUG-RESISTANT NEISSERIA GONORRHOEAE has the highest number of treat estimate with about 550 000 infections, and with a Resistant infection that is increasing.
Neisseria gonorrhoeae (Gram-negative bacteria) causes gonorrhea, a sexually transmitted disease (STD) that can result in life-threatening ectopic pregnancy and infertility, and can increase the risk of getting and giving HIV. treatment with ceftriaxone for N. gonorrhoeae infections is highly effective, but there is growing concern about antibiotic resistance.
Gonorrhea has quickly developed resistance to all but one class of antibiotics, and half of all infections are resistant to at least one antibiotic. Tests to detect resistance are not available at time of treatment. Gonorrhea spreads easily. Some men and most women do not have symptoms and may not know they are infected, increasing spread. Untreated gonorrhea can cause serious and permanent health problems in women and men, including ectopic pregnancy and infertility, and can spread to the blood resulting in cardiovascular and neurological problems.
Problem Statement
We will be focusing on a species called Neisseria gonorrhoeae, the bacteria which cause gonorrhoea. Gonorrhoea is the second most common sexually transmitted infection (STI) in Europe, after chlamydia. Rates of gonorrhoea infection are on the rise, with a 26% increase reported from 2017–2018 in the UK. Many people who are infected (especially women) experience no symptoms, helping the disease to spread. If the infection is left untreated, it can lead to infertility in women, and can occasionally spread to other parts of the body such as your joints, heart valves, brain or spinal cord.
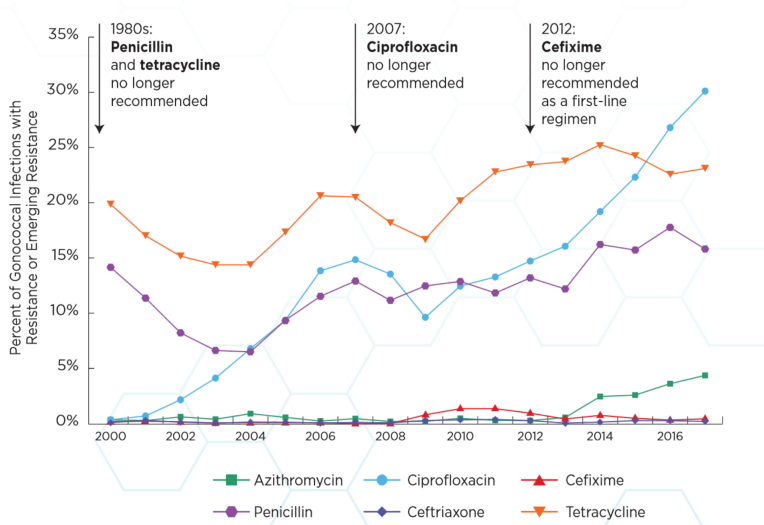 |
|---|
| The rates of resistance to different antibiotics. Resistance of these bacteria to antibiotics is rising over time, making infections hard to treat. Image is from this report |
In the past, patients were treated with an antibiotic called ciprofloxaxcin. Doctors had to stop using this antibiotic because resistance to the drug became too common, causing treatments of infections to fail. Until very recently, the recommended treatment was two drugs — ceftriaxone and azithromycin. Azithromycin was removed from recommendations because of concern over rising resistance to the antibiotic. In February 2018, the first ever reported case of resistance to treatment with ceftriaxone and azithromycin, as well as resistance to the last-resort treatment spectinomycin, was reported. Currently in the UK, patients are only treated with ceftriaxone.
In this blog, we will look at machine learning algorithms for predicting resistance to ciprofloxacin. we will look into what your model has learned, and whether this fits with our existing knowledge of antibiotic resistane, also we will examine how much we’d benefit from collecting more samples, and explore the impact of genetic relatedness on accuracy measures. Some further tasks could be, to explore more hyper-parameters and try to build more accurate models, to try some other model types, and to try using the unitigs to predict resistance to another antibiotic included in metadata.csv
Dataset
For this exercise, we have genome sequence and antibiotic resistance data gathered from different publicly available sources. If you’d like to learn more about the collection, an interactive view of the data can be accessed here.
For this analysis, we’re using unitigs, stretches of DNA shared by a subset of the strains in our study. Unitigs are an efficient but flexible way of representing DNA variation in bacteria. If you’d like to learn more about unitigs, and how this dataset was constructed, have a look at this paper.
The full dataset consists of 584,362 unitigs, which takes a long time to train models on, so for this exercise we will be using a set that has been filtered for unitigs associated with resistance.
Training data
For this exercise, we worked with unitigs, segments of DNA shared by strains in our collection. These are produced by taking the DNA from different bacteria that we’ve pieced together after sequencing their genomes, then breaking it into different 31-character words. These words are then assembled into a De Bruijn graph (DBG). This graph is pieced together by identifying words that overlap internally, and are present in the same samples.
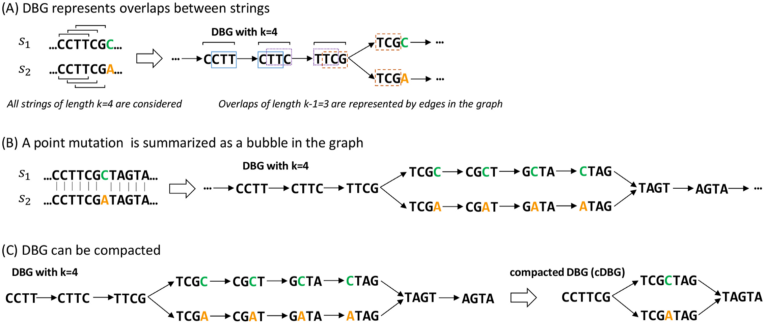 |
|---|
| Image is taken from this paper. Please refer to it for more detail |
This process allows us to represent the similarities and differences between these different bacteria in an efficient way. The differences can be individual mutations, pieces of DNA that have been inserted or deleted, or other genetic elements called plasmids, which can pass between bacteria and spread antibiotic resistance.
Usually when working with unitigs, the number of variables may range between 0.5–5 million, but for the purpose of the exercise I first ran an association study to identify unitigs that were significantly associated with resistance to make the analysis run faster.
The filtering resulted in 8,873 unitigs strongly associated with ciprofloxacin resistance and 515 unitigs significantly associated with azithromycin resistance, for a dataset containing 3,971 samples.
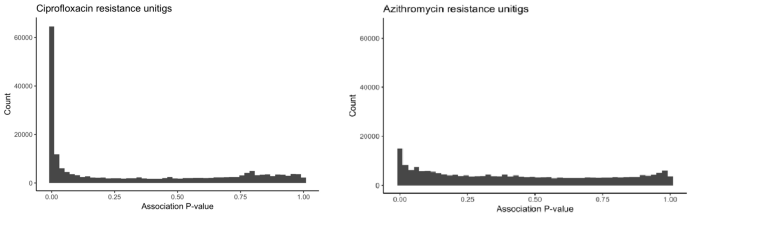 |
|---|
| The distribution of p values for unitigs with ciprofloxacin and with azithromycin |
Predicting Methods
We will start will building some basic models for ciprofloxacin resistance. This resistance pattern can mostly be explained by a single mutation, so is likely to be impacted by the amount of noise each method incorporates. So the data must be clean, accurate and standardized.
After having our data organized, we can start fitting models. First we will try an elastic net logistic regression, then we will try a support vector machine, and after that XGBoost.
Results
We used K Fold cross validation, with k=5, the following graph compares results from the different predictors
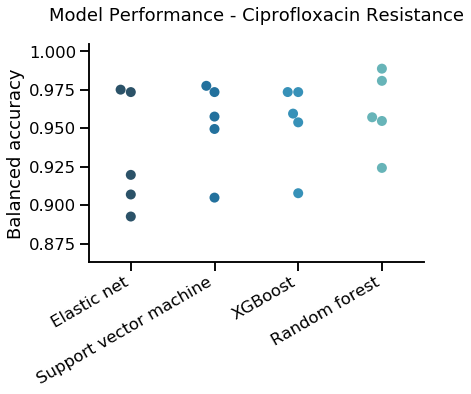 |
|---|
| K Fold cross validation for different predictors for ciprofloxacin |
By exploring what the RF model has learned, we found that Top negative predictors are:
‘GTGCGACAGCAAAGTCCAAACCAGCGTCCCCGCC’
‘GCGCAGCCGCAAATCTTGTTTTCCCATTCCGCC’
‘GCCGAAATATTCCGTCAGCAGTTTTTCCCCC’
‘GCGGCGGCAGGGGTAGGTACGGTCGTTTTGGGCAGGGG’
‘ATCGATTGCGCTTCAAGGCCCTGCATGTGCCT’
and top positive predictors are:
‘GGCATCCCGAAGCCGAATACGGCAACGGCAAGCG’
‘GCGGCGCAGGGCGATGATTTGGTTTTCGTCC’
‘ATGACCGAACAACACTTTACCGAACAAATCAAATCCCTAATCGACAGCTTAAAAACCAT’
‘CGTCTGAAAAAACACAATATCGATGTCTATATTATGAGCGGCGA’
‘GTATTCGCGGATATATGCCTGATCCAGCCCGAGGCACAA’
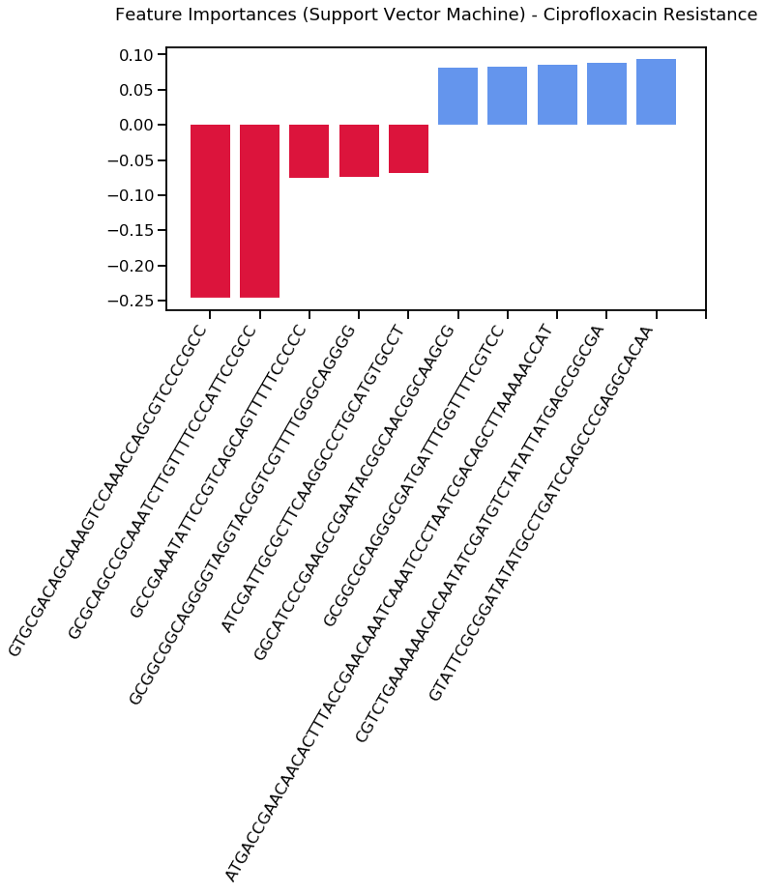 |
|---|
| Most important unitigs associated with ciprofloxacin using Support vector machine |
Learning curve for RF:
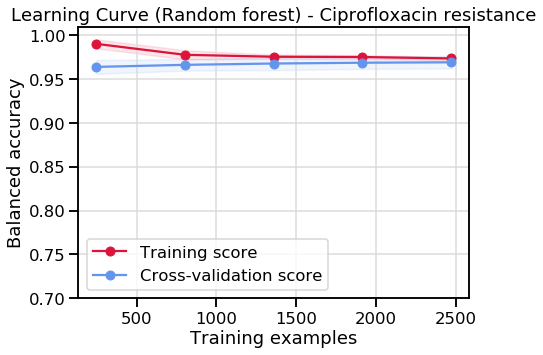 |
|---|
| Learning curve associated with ciprofloxacin resistance using random forest |
You can take the unitigs from above, and input one into this search algorithm: https://www.uniprot.org/blast or https://card.mcmaster.ca/analyze/blast to see if it comes from a known protein or resistance mechanism. The search has to be formatted like this for CARD:
>sequence
[unitig]
and you will need to choose the BLASTN option for the query to be processed correctly.
If you want to look into the biology more, try this link, to see where the unitigs can be found in a publicly available collection of genomes.
Discussion
The random forest has found a nice solution to the problem, but the SVM appears to have over-fit.
By seeing at how much performance improves as we include more of our sample set, and whether performance gains have levelled of. Both models don’t look like they’d particularly benefit from the collection of more samples.
Ciprofloxacin resistance can usually be predicted in any species by looking for one or two mutations that cause the vast majority of resistance. In this sense, they are easy models to build, but ML models can sometimes have trouble building a model that isn’t cluttered with extraneous information.
When I looked into what the models had learned, I was disappointed to see that while both models were quite accurate, the major mutation that we know drives resistance hadn’t been chosen as a top feature by either of these models, suggesting the models had found an alternative solution to accurately predicting resistance that didn’t involve the actual resistance mechanism.
This is a relatively common problem in ML in this area — there are so many variables, and so few samples, that spurious associations can form in the data. It’s possible this high performance is due to the model identifying unitigs that flag major families of resistant strains rather than real resistance mechanisms. The best way of diagnosing this would be to see where the unitigs fall on a phylogenetic tree of the isolates.
References
The dataset and code could be found at this link
- https://www.kaggle.com/mmadkour/predicting-antibiotic-resistance-infections?scriptVersionId=40020048
Read more about antibiotic resistance here:
- The impact of antibiotic resistance on modern medicine
- Phage therapy, a possible solution to the antibiotic resistance crisis
- Should machine learning algorithms guide antibiotic prescribing?
comments powered by Disqus